
- Generally amino acids are the end products of the digestion of dietary proteins. In humans, the degradation of ingested proteins to their constituent amino acids occurs in the gastro-intestinal tract with the help of respective proteolytic enzymes.
- The resulting mixture of free amino acids is transported into the epithelial cells lining the small intestine, through which the amino acids enter the blood capillaries in the villi and travel to the liver.
- Some of the non-essential amino acids are biosynthesized in our body as well.
- In our body, unlike carbohydrates and lipids, oxidative degradation of amino acids is least preferred for the generation of energy.
- In animals, amino acids undergo oxidative degradation in three different metabolic circumstances:
- During the normal synthesis and degradation of cellular proteins, some amino acids that are released from protein breakdown and are not needed for new protein synthesis undergo oxidative degradation.
- When a diet is rich in protein and the ingested amino acids exceed the body’s needs for protein synthesis, the surplus is catabolized because amino acids cannot be stored.
- During starvation or in uncontrolled diabetes mellitus, when carbohydrates are either unavailable or not properly utilized, cellular proteins are used as fuel.
- Most of the amino acids are metabolized in liver cells. Amino group of amino acids provide nitrogen for the synthesis of some biomolecules such as nitrogenous bases. Rest of the nitrogen in the form of ammonia is converted into urea in liver cell and is excreted. It is because if ammonia accumulates in cells or blood, it results in harmful consequences since it is toxic.
Steps in the catabolism of amino acids:
- Transfer of amino group from amino acids to α-ketoglutarate (Transamination):
- This process occurs in the cytosol of liver cells where α-amino group from all 19 amino acids (except glutamate) is transferred to the α-carbon atom of α-ketoglutarate to form L-glutamate and corresponding keto-acid analogue of the amino acid.
- This reaction is called transamination and is catalyzed by enzymes called aminotransferases or transaminases
- Since, α-ketoglutarate becomes aminated and α-amino acid is deaminated, there is no net deamination.
- The purpose of transamination reactions is to collect the amino groups from many different amino acids in the form of L-glutamate, which then functions as an amino group donor for biosynthetic pathways or excretion pathways that lead to the elimination of nitrogenous waste products.
- Removal of amino group from L-glutamate (Deamination):
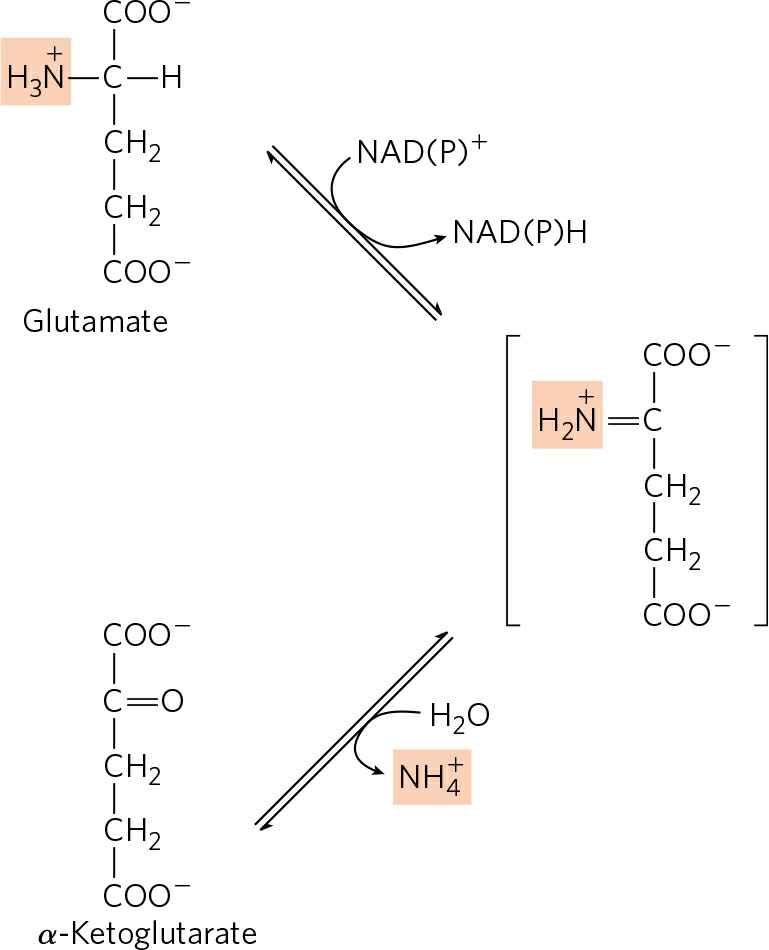
- L-glutamate formed in the cytosol of hepatocytes is now transported into mitochondria, where it undergoes oxidative deamination.
- L-glutamate releases its amino group into the mitochondrial matrix catalyzed by L-glutamate dehydrogenase.
- The combined action of aminotransferase and glutamate dehydrogenase is referred to as trans-deamination.
- A few amino acids bypass this trans-deamination pathway and undergo direct oxidative deamination.
- Thus released amino group is channeled into urea cycle for excretion.
- The α-ketoglutarate formed from glutamate dehydrogenase can be used in the citric acid cycle and for glucose synthesis.
Transportation of ammonia from other organs (except skeletal muscles) to the liver:
- In many tissues, including the brain, some processes such as nucleotide degradation generate ammonia, which is quite toxic.
- This free ammonia has to be converted into a non-toxic compound before export from extra-hepatic tissues into the blood and transport to the liver or kidneys.
- For this transport function, glutamate, critical to intracellular amino group metabolism, is replaced by L-glutamine.
- The free ammonia produced in tissues is combined with L-glutamate to yield L-glutamine by the action of L-glutamine synthetase.
- L-glutamine which is a nontoxic transport form of ammonia is then transported to the liver through blood and then enters into the mitochondria of liver cell.
- Inside the mitochondria, it releases its amide nitrogen as ammonium ion. Glutaminase converts L-glutamine to L-glutamate and NH4+.
- Ammonia from all sources is disposed of by urea synthesis.

Transportation of ammonia from skeletal muscles to the liver:
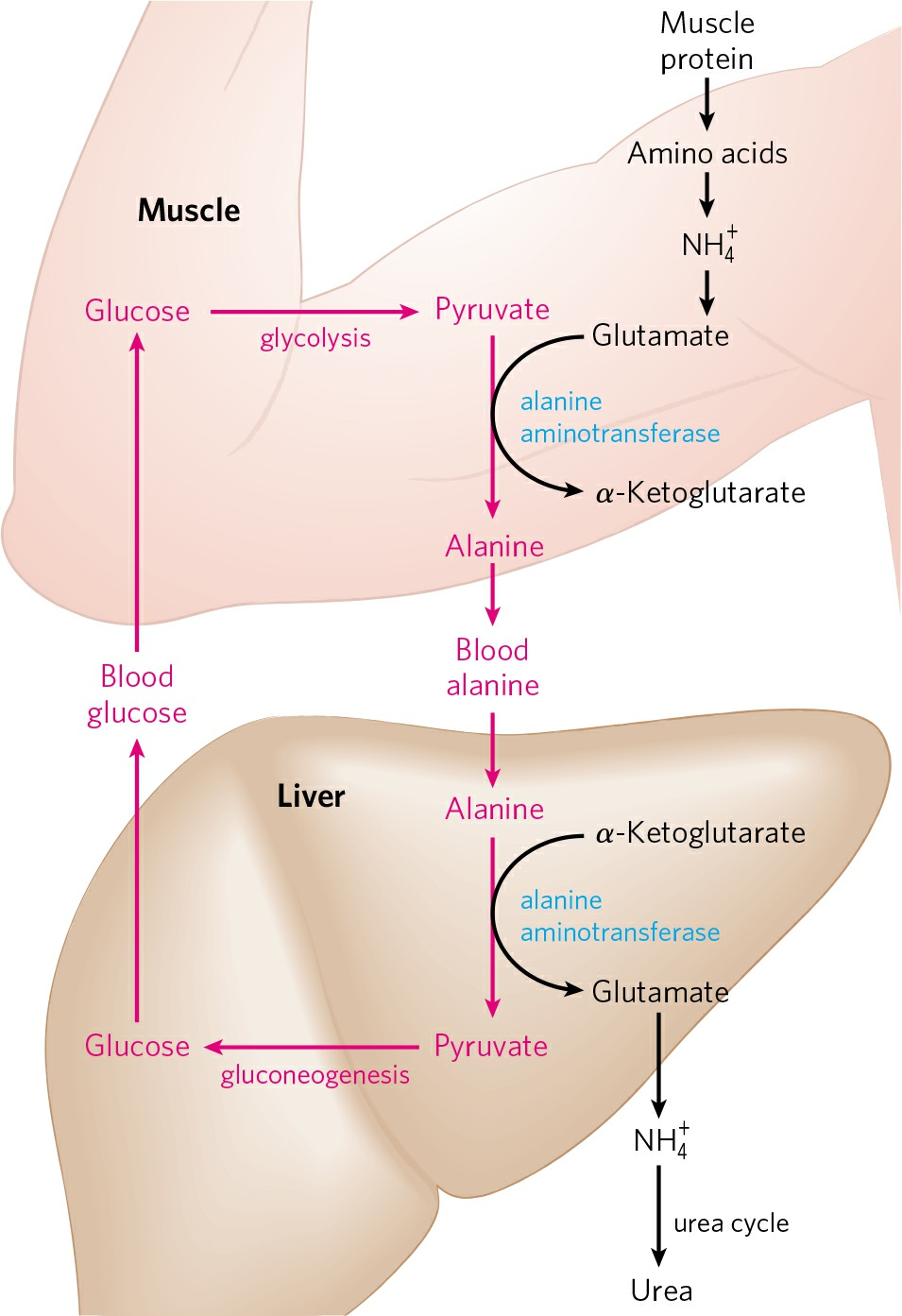
- From muscle, amino group is transported to liver in the form of L-alanine via a pathway called glucose-alanine cycle.
- At first, L-glutamate is formed in muscle cells by transamination (same as in cytosol of hepatocytes).
- L-glutamate then transfers its α-amino group to pyruvate, a readily available product of muscle glycolysis to form alanine. This reaction is brought about by an enzyme called alanine aminotransferase.
- L-alanine thus formed passes into the blood and travels to the liver.
- In the cytosol of hepatocytes, L-alanine again transfers its amino group to α-ketoglutarate to form L-glutamate and free L-alanine catalyzed by alanine aminotransferase.
- L-glutamate can then enter the mitochondria, where glutamate dehydrogenase reaction releases NH4+, or can undergo transamination with oxaloacetate to form aspartate, another nitrogen donor in urea synthesis.
Trans-deamination (Metabolism of amino acids) and transport of ammonia from other organs to liver for detoxification