
- Animals require pathways for nitrogen excretion that minimizes toxicity in the body.
- If not reused for the synthesis of new amino acids or other nitrogenous products, amino groups are channeled into a single excretory end product. The end product can either be ammonia, urea or uric acid.
- Most of the aquatic species, such as the bony fishes, are ammonotelic (excreting amino nitrogen as ammonia).
- Most terrestrial animals are ureotelic (excreting amino nitrogen in the form of urea) whereas birds and reptiles are uricotelic (excreting amino nitrogen as uric acid).
- In ureotelic organisms, the ammonia deposited in the mitochondria of hepatocytes (liver cells) is converted to urea through the urea cycle.
You may also want to see: Trans-deamination (Metabolism of amino acids) and transport of ammonia from other organs to liver for detoxification
Urea Cycle:
- This pathway was discovered by Hans Krebs and Kurt Henseleit in 1932.
- Proteins and amino acids are metabolized to form urea.
- The nitrogen of amino acid, converted to ammonia, is toxic to body. It is converted to urea and detoxified.
- Urea production occurs almost exclusively in the liver and is the fate of most of the ammonia channeled there.
- The urea thus formed then passes into the bloodstream and to the kidneys and is excreted into the urine. As such urea accounts for 80-90% of nitrogen containing substances excreted in urine.
Steps of urea cycle:
- Urea synthesis is a five step cyclic process, with five distinct enzymes. The first two enzymes are present in mitochondria while the rest are located in cytosol.
- Hence, urea cycle begins inside liver mitochondria, but three of the subsequent steps take place in the cytosol; the cycle thus spans two cellular compartments.
- Urea has two amino (-NH2) groups, one derived from NH3 from the mitochondrial matrix and the other from aspartate (generated in mitochondria by transamination and transported to cytosol).
- Carbon atom in urea is obtained from CO2 (as HCO3–) produced by mitochondrial respiration.
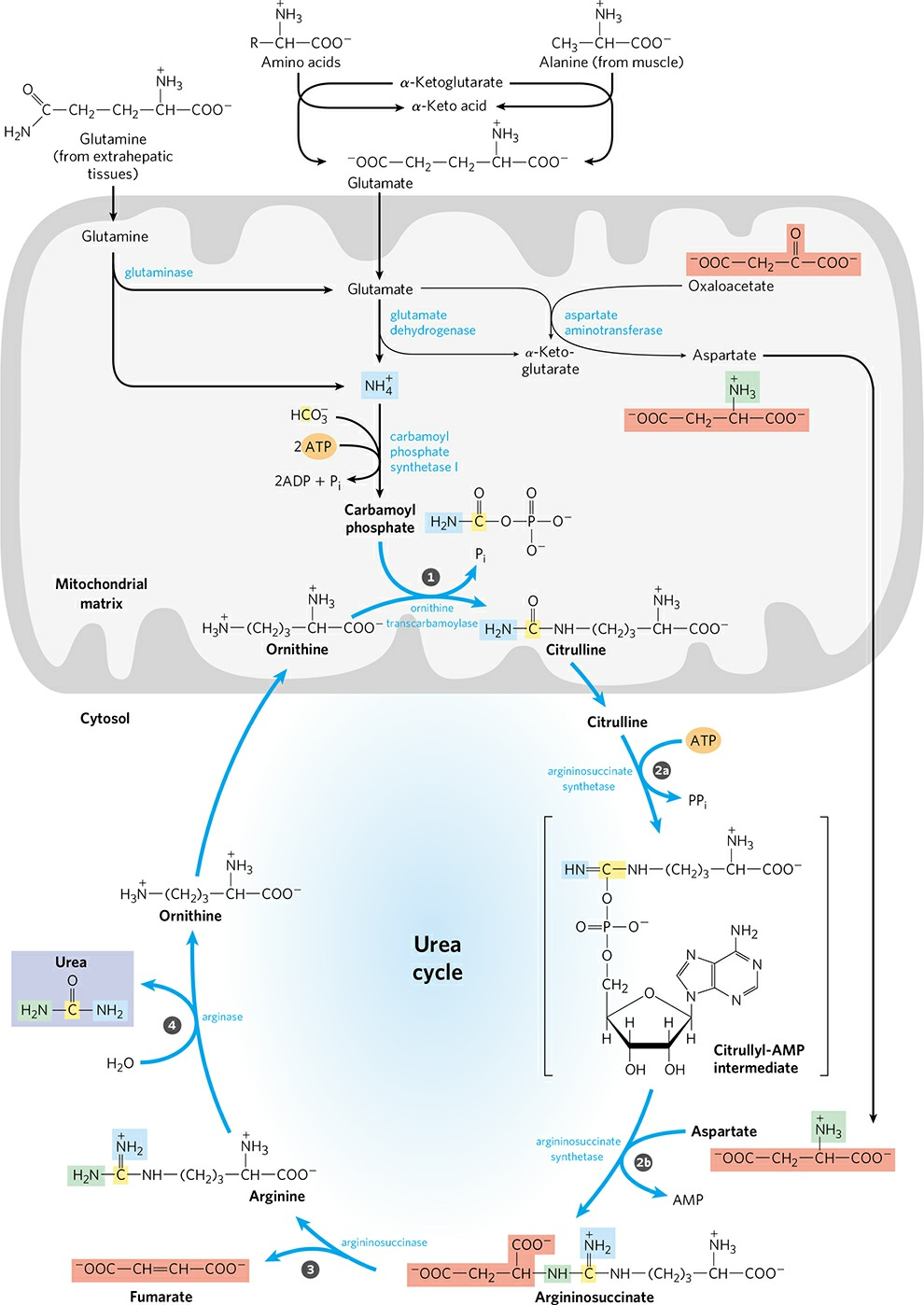
- Five enzymatic steps of urea cycle are as follows:
- Synthesis of carbamoyl phosphate:
- NH4+ generated in liver mitochondria is used, together with CO2 (as HCO3–) produced by cellular respiration, to form carbamoyl phosphate in the matrix.
- This ATP-dependent condensation reaction consumes two ATP and is irreversible and rate limiting. It is catalyzed by a regulatory enzyme, carbamoyl phosphate synthetase I (CPS I), that requires N-acetyl glutamate for its activity.
- The mitochondrial form of CPS I is distinct from the cytosolic form (CPS II), which has a separate function in pyrimidine biosynthesis.
- CPS II accepts amino group from glutamine and doesn’t require N-acetyl glutamate for its activity.
- Carbamoyl phosphate, which functions as an activated carbamoyl group donor, now enters the urea cycle.
2. Formation of Citrulline:
- Carbamoyl phosphate donates its carbamoyl group to ornithine to form citrulline catalyzed by ornithine transcarbomoylase.
- Ornithine plays a role resembling to that of oxaloacetate in citric acid cycle. Ornithine and citrulline are basic amino acids. They are never found in protein structure due to lack of codons.
- Citrulline produced in this reaction is transported from mitochondrial matrix to cytosol by a transporter system.
- Synthesis of Argininosuccinate:
- The second amino group of urea is incorporated in this reaction.
- Argininosuccinate synthetase condenses citrulline with aspartate to produce argininosuccinate. Condensation reaction occurs between the amino group of aspartate and the ureido (carbonyl) group of citrulline. A citrullyl-AMP intermediate is formed.
- This step requires ATP and which is cleaved to AMP and pyrophosphate (PPi). The latter is immediately broken down to inorganic phosphate (Pi).
- Cleavage of Argininosuccinate:
- Argininosuccinate is cleaved by argininosuccinase to give free Arginine and Fumarate which the only reversible step in the urea cycle.
- Arginine is the immediate precursor for urea.
- Fumarate thus formed enters the mitochondria to join the pool of TCA cycle intermediates connecting TCA cycle, gluconeogenesis etc.
- Formation of urea:
- In the last reaction of urea cycle, the cytosolic enzyme arginase, cleaves arginine to yield urea and ornithine. Arginase is activated by Co2+ and Mn2+.
- Ornithine so regenerated, is transported into mitochondria to initiate another round of urea cycle.
- The urea thus formed then passes into the bloodstream and to the kidneys and is excreted into the urine.
- Ornithine and lysine compete with arginine (competitive inhibition).
- Arginase is mostly found in the liver, while the rest of the enzymes of urea cycle are also present in other tissues.
- For this reason, arginine synthesis may occur to varying degrees in many tissues but only liver can ultimately produce urea.
Metabolic disorders of Urea Cycle:
- A urea cycle disorder (UCD) is an inherited disease that affects how the body removes the waste that is made from breaking down protein.
- It results in hyperammonemia, encephalopathy, respiratory alkalosis.
- UCDs affect about 1 in 35,000 newborns.
- UCDs occur because of deficiency of some urea cycle enzymes as follows:
- Carbamoyl phosphate synthetase I (CPS I) Deficiency:
- It is an autosomal recessive disorder.
- Complete lack of CPS I causes hyperammonemia in newborns. It is a very severe urea cycle disorder and even after treatment and recovery, they are at chronic risk of bouts of hyperammonemia. Partial or mild CPS I deficiency can cause symptoms at any stage of life, especially in response to stress or infection.
- It has two forms; early onset and delayed onset forms.
- Early onset is characterized by moderate to severe cerebral damage and hyperamonemic coma in neonatal. CPS I is totally absent in it.
- Delayed onset is due to partial deficiency of CPS I.
- Ornithine transcarbamylase (OTC) Deficiency:
- OTC deficiency is inherited in an X-linked recessive manner; males are more commonly affected than females.
- Females are at lesser risk of OTC deficiency as they have 2 X chromosomes and chances of having one functional OTC gene are higher. Only about 15% of females show symptoms of OTC deficiency and develop hyperammonemia, necessitating chronic medical management.
- The severe form of OTC deficiency occurs in some affected males anywhere between 24 hours to a few days after birth, usually following a protein feeding. Initial symptoms may include refusal to eat, poor suck, vomiting, progressive lethargy, and irritability.
- Argininosuccinate synthetase Deficiency or Citrullinemia Type I:
- When it is deficient, either fully or partially ammonia can build up and cause different pathological conditions (hyperammonemia).
- They also have abnormally high levels of citrulline in blood.
- Increased level of ammonia in cerebrospinal fluid includes excessive vomiting, anorexia, irritability, lethargy, seizure and respiratory distress.
- Argininosuccinase (ASL) Deficiency:
- It is an autosomal recessive disorder.
- ASL deficiency is the second most common UCD with the prevalence of 1 in 70,000 live births.
- This disorder is characterized by rapid hyperammonemia in newborns. It causes chronic hepatic enlargement. Enlarged hepatocytes may lead to fibrosis.
- Arginase (ARG) Deficiency :
- It is an inherited metabolic disease, where a deficiency of the enzyme arginase causes a buildup of the enzyme arginine and ammonia in blood.
- This defect does not cause rapid onset of hyperammonemia but some patients experience severe symptoms such as progressive spasticity (condition in which muscles stiffen or tighten, preventing normal fluid movement) and retarded growth, intellectual disability, problems with balance and coordination.
- Carbamoyl phosphate synthetase I (CPS I) Deficiency:
Nitrogen excretion through Urea cycle and associated disorders
Reference:
- Lehninger, A. L., Nelson, D. L., & Cox, M. M. (2000). Lehninger principles of biochemistry. New York: Worth Publishers